¶ Microglia[1]
¶ Introduction
Microglia is an important component in the neurobiology of neurodegenerative diseases. This page provides detailed information about its structure, function, and role in disease processes.
¶ Overview
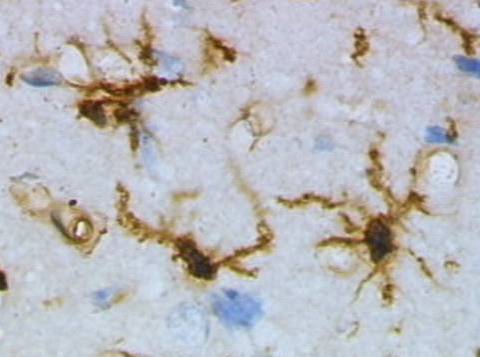
microglia cells visualized with lectin staining. Image source: Wikimedia Commons (Public Domain).
Microglia[1] are the resident immune cells of the [central nervous system], constituting approximately 10–15% of the total brain cell population.
Derived from embryonic yolk sac progenitors, microglia colonize the brain during early development and self-renew throughout life without contribution from peripheral bone
marrow-derived monocytes ([Prinz et al., 2019). These highly dynamic cells continuously survey the brain parenchyma, maintaining homeostasis through phagocytosis of debris and dead
cells, synaptic pruning, release of neurotrophic factors, and immune surveillance. In neurodegenerative diseases — including [Alzheimer3's disease], Parkinson's disease, ALS, and frontotemporal dementia — microglia adopt altered activation states that can be both protective
(clearing [protein aggregates) and detrimental (driving chronic [neuroinflammation. Genome-wide association studies have identified numerous microglial genes as risk loci for
neurodegeneration, including TREM2, CD33, SPI1, and ABI3, underscoring the central role of microglia in disease pathogenesis ([Xu & bhatt, 2023).
¶ Development and Origin
Microglia[1] are unique among brain cells in their myeloid origin. They emerge from early erythromyeloid precursor cells (EMPs) in the extraembryonic yolk sac at approximately embryonic day 7.5 (E7.5 in mice), subsequently colonizing the developing brain rudiment by E9.5 ([Ginhoux et al., 2010). Unlike other tissue macrophages, microglia are maintained throughout life by local self-renewal rather than replacement from circulating monocytes.
Microglia[1]l development proceeds through three distinct phases:
- Early microglia (until E14): Initial colonization and proliferation
- Pre-microglia (E14 to the first postnatal weeks): Acquisition of a ramified morphology and homeostatic gene expression
- Adult microglia (from several weeks postnatal onward): Mature surveillance state
These developmental transitions require continuous instructive signaling from the brain environment, particularly transforming growth factor-β (TGF-β) signaling and colony-stimulating factor-1 receptor ([CSF1R) signaling. CSF1R signaling, driven by CSF1 and IL-34 ligands, is essential for microglial survival, proliferation, and differentiation ([Prinz et al., 2019).
¶ Morphology and Distribution
Microglia[1] exhibit remarkable morphological plasticity that reflects their functional state:
- Ramified (homeostatic) state: In the healthy brain, microglia display small cell bodies with numerous highly branched processes that continuously extend and retract to survey their territory. Each microglial cell monitors a discrete territory of approximately 50,000 µm³, and their processes can scan the entire brain parenchyma within a few hours.
- Activated (reactive) state: In response to injury, infection, or disease signals, microglia retract their processes and adopt an amoeboid morphology with enlarged cell bodies, enhanced phagocytic capacity, and altered secretory profiles.
- Rod-shaped microglia: Found in response to diffuse brain injury, these elongated cells align along neuronal dendrites and may play a role in synaptic stripping.
Microglia[1] are unevenly distributed throughout the brain, with the highest densities in the hippocampus, substantia nigra, basal ganglia, and cortex, and lower densities in white matter and the cerebellum.
¶ Functions in Brain Homeostasis
¶ Immune Surveillance
Microglia[1] serve as the brain's first line of immune defense. Their processes continuously monitor the parenchyma for signs of infection, injury, or abnormal protein accumulation. They express an array of pattern recognition receptors including Toll-like receptors (TLR4, TLR2), TREM2, and scavenger receptors that detect pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Upon detecting danger signals, microglia rapidly mobilize to the site of injury and initiate an inflammatory response.
¶ Synaptic Pruning
During development, microglia play an essential role in sculpting neural circuits by eliminating excess synaptic connections through [complement]-mediated phagocytosis ([Schafer et al., 2012). Weaker or less active synapses are tagged with complement proteins C1q and C3, which are recognized by the microglial complement receptor CR3, triggering engulfment. This process is also mediated by additional ligand-receptor pathways including phosphatidylserine–TREM2, CX3CL1–CX3CR1, and Gas6–MerTK.
Critically, this developmental pruning pathway can be pathologically reactivated in neurodegenerative diseases, leading to aberrant complement-mediated synapse loss — a major driver of cognitive decline in [Alzheimer[3]'s disease] ([Hong et al., 2016).
¶ Neurotrophic Support
Microglia[1] secrete growth factors that support neuronal survival and function, including brain-derived neurotrophic factor (BDNF), insulin-like growth factor-1 (IGF-1), and nerve growth factor (NGF). They also release anti-inflammatory cytokines such as IL-10 and TGF-β that maintain a neuroprotective environment. Through IGF-1 secretion, microglia support oligodendrocytes survival and [myelination].
¶ Phagocytosis and Debris Clearance
Microglia[1] are the primary phagocytes of the CNS, clearing apoptotic cells, myelin debris, and aberrant protein aggregates including [amyloid-β], alpha-synuclein, and TDP-43. This clearance function is essential for maintaining tissue homeostasis and is regulated by receptors including TREM2, TAM receptors (Tyro3, Axl, MerTK), and scavenger receptors.
¶ Microglial States in Disease
¶ Disease-Associated Microglia[1] (DAM)
Single-cell RNA sequencing has revealed that microglia adopt a distinct transcriptional state in neurodegenerative diseases, termed disease-associated microglia (DAM) (Keren-Shaul et al., 2017). DAM activation occurs through a two-stage process:
¶ Inflammatory Activation
Chronic microglial activation in neurodegenerative diseases leads to sustained release of pro-inflammatory mediators including [IL-1β], [TNF-α], IL-6, reactive oxygen species, and nitric oxide. The NLRP3 inflammasome], a multi-protein complex activated by [amyloid-β], damaged mitochondria, and other danger signals, is a major driver of microglial IL-1β production and pyroptotic cell death ([Heneka et al., 2018). This chronic neuroinflammatory state exacerbates [neuronal death], disrupts the blood-brain barrier, and amplifies protein aggregation.
¶ Role in Specific Neurodegenerative Diseases
¶ Alzheimer[3]'s Disease
In [Alzheimer[3]'s disease], microglia play a complex dual role:
- Protective functions: Microglia[1] cluster around [amyloid-β plaques] and form a barrier that limits plaque expansion and fibril spread. TREM2 signaling is essential for this plaque-associated microglial response, and TREM2 loss-of-function variants increase AD risk approximately 3-fold. TREM2 also protects against [complement-mediated synaptic loss] by binding C1q ([Bhatt et al., 2023).
- Detrimental functions: Chronically activated microglia release neurotoxic inflammatory mediators, phagocytose viable synapses via aberrant complement tagging, and impair [glymphatic clearance] of [amyloid-β] and tau].
Genetic studies have identified numerous microglial genes as AD risk loci, including TREM2, CD33, APOE(https://pubmed.ncbi.nlm.nih.gov/35379992/)).
¶ Parkinson's Disease
Microglia[1]l activation in Parkinson's disease contributes to the progressive loss of dopaminergic neurons in the substantia nigra:
- Aggregated alpha-synuclein activates microglia through TLR2 and TLR4, triggering NF-κB-mediated inflammatory signaling
- Microglia[1]l complement system activation tags dopaminergic synapses for elimination
- Impaired microglial phagocytosis of alpha-synuclein contributes to its spread and propagation
- LRRK2 mutations, the most common genetic cause of PD, alter microglial inflammatory responses
¶ Amyotrophic Lateral Sclerosis
In ALS, microglia exhibit stage-dependent responses that shift from neuroprotective to neurotoxic as disease progresses. Microglia[1] contribute to motor neuron degeneration through NLRP3 inflammasome activation, release of excitotoxic factors, and impaired clearance of TDP-43 and SOD1 aggregates. Single-nucleus RNA sequencing has revealed spatiotemporal dynamics of DAM in ALS, with region-specific microglial activation patterns correlating with motor neuron vulnerability ([Chen et al., 2024).
¶ Frontotemporal Dementia
Loss-of-function mutations in GRN (progranulin), a major FTD risk gene, cause severe microglial dysfunction including impaired lysosomal function, excessive complement activation, and altered synaptic pruning. TREM2 mutations also cause a form of FTD (Nasu-Hakola disease), highlighting the critical role of microglial function in frontotemporal neurodegeneration.
¶ TREM2: A Master Regulator of Microglial Function
TREM2 (Triggering Receptor Expressed on Myeloid cells 2) is a transmembrane receptor expressed predominantly on microglia that has emerged as a master regulator of microglial activation and a major genetic risk factor for neurodegeneration. TREM2 functions include:
- Phagocytosis enhancement: TREM2 binds lipids, APOE is elevated in AD and serves as a biomarker of microglial activation ([Shi et al., 2025).
¶ Therapeutic Strategies Targeting Microglia
Microglia[1] represent increasingly attractive therapeutic targets for neurodegenerative diseases:
- TREM2 agonists: Activating antibodies and small molecules that enhance TREM2 signaling are in development to promote protective microglial responses. AL002 (an anti-TREM2 antibody) has entered clinical trials for AD.
- NLRP3 inflammasome inhibitors: Compounds targeting the NLRP3 inflammasome (e.g., MCC950, dapansutrile) aim to reduce harmful neuroinflammation[2].
- CSF1R modulators: CSF1R inhibitors (e.g., PLX5622) can deplete microglia, allowing repopulation with a reset homeostatic phenotype. CSF1R agonists may enhance microglial survival and function.
- Complement inhibitors: Blocking aberrant complement-mediated synaptic pruning, particularly the C1q–C3–CR3 axis, may preserve synapses in neurodegeneration.
- Microglia[1]l reprogramming: Approaches to shift microglia from pro-inflammatory to neuroprotective states, including enhancing lysosomal acidification and modulating lipid metabolism.
- Anti-CD33 therapies: Reducing CD33 signaling enhances microglial phagocytosis of amyloid-β.
¶ Current Research
¶ Key Research Directions
- Single-cell atlases: Large-scale single-cell and spatial transcriptomic studies are mapping the full diversity of microglial states across human neurodegenerative diseases and brain regions
- Microglia[1]l metabolism: Understanding how energy metabolism (glucose, lipid) shapes microglial function and dysfunction
- Sex differences: Investigating why microglial activation patterns and neuroinflammatory responses differ between males and females
- Microglia[1]l extracellular vesicles: Studying how microglial exosomes contribute to protein aggregate propagation
- Human-specific biology: Identifying microglial features unique to humans that are not captured in mouse models
¶ Brain Atlas Resources
- Allen Human Brain Atlas: Microglia expression search
- Allen Mouse Brain Atlas: Microglia search
- Allen Cell Type Atlas: Transcriptomic cell type reference
- BrainSpan Developmental Transcriptome: Microglia developmental expression
¶ See Also
¶ External Links
¶ Background
The study of Microglia has evolved significantly over the past decades. Research in this area has revealed important insights into the underlying mechanisms of neurodegeneration and continues to drive therapeutic development.
Historical context and key discoveries in this field have shaped our current understanding and will continue to guide future research directions.
¶ References
- [Prinz, M., Jung, S., & Priller, J. (2019). Microglia[1] biology: one century of evolving concepts. Cell, 179(2), 292-311. [PubMed)]https://pubmed.ncbi.nlm.nih.gov/30206328/)
- [Ginhoux, F., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 330(6005), 841-845. [PubMed)]https://pubmed.ncbi.nlm.nih.gov/21030899/)
- [Keren-Shaul, H., et al. (2017). A unique microglia type associated with restricting development of Alzheimer[3]'s disease. Cell, 169(7), 1276-1290. [PubMed)]https://pubmed.ncbi.nlm.nih.gov/28602351/)
- PubMed)](https://pubmed.ncbi.nlm.nih.gov/30116888/
- [Hong, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer[3] mouse models. Science, 352(6286), 712-716. [PubMed)]https://pubmed.ncbi.nlm.nih.gov/27033548/)
- PubMed)](https://pubmed.ncbi.nlm.nih.gov/22632727/
- [Bellenguez, C., et al. (2022). New insights into the genetic etiology of Alzheimer[3]'s disease and related dementias. Nature Genetics, 54(4), 412-436. [PubMed)]https://pubmed.ncbi.nlm.nih.gov/35379992/)
- [Xu, Y., et al. (2023). Microglia[1] in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduction and Targeted Therapy, 8, 443. [PubMed)]https://pubmed.ncbi.nlm.nih.gov/37735487/)
- [Bhatt, D. L., et al. (2023). TREM2 receptor protects against complement-mediated synaptic loss by binding to complement C1q during neurodegeneration. Immunity, 56(8), 1794-1808. [PubMed)]https://pubmed.ncbi.nlm.nih.gov/37442133/)
- [Chen, W., et al. (2024). Single-nucleus RNA sequencing reveals the spatiotemporal dynamics of disease-associated microglia in amyotrophic lateral sclerosis. Research, 7, 0548. [Link)]https://spj.science.org/doi/10.34133/research.0548)
- [Human Microglia[1] Atlas Consortium. (2025). The Human Microglia[1] Atlas (HuMicA) unravels changes in disease-associated microglia subsets across neurodegenerative conditions. Nature Communications, 16, 1234. [Link)]https://www.nature.com/articles/s41467-025-56124-1)
- [Colonna, M., & Wang, Y. (2016). TREM2 variants: new keys to decipher Alzheimer[3]'s disease pathogenesis. Nature Reviews Neuroscience, 17(4), 201-207. [PubMed)]https://pubmed.ncbi.nlm.nih.gov/26911434/)## See Also
-
- Proteins Index## External Links